
DTI?
화학 물질(Drug)과 그 물질의 표적이 되는 단백질(Drug target) 사이의 상호작용(DTI) score를 예측함을 목표로 하고 실습을 진행해 보자.
데이터 형식
주어질 데이터의 형태는 다음과 같다.
- 단백질 ← peptide sequence
- Drug ← SMILES 방식 sequence
기대 효과

임상 전에 약물이 특정 단백질과 상호작용하는 수치를 미리 예측할 수 있고 연구 개발 단계에서 소요되는 시간을 획기적으로 줄일 수 있을 것이다. 이렇게 가상환경에서의 실험 방법을 In-Silico라고 하며 앞으로 주목받을 차세대 연구 개발 항목임에 틀림없다.
데이터 준비
https://www.nature.com/articles/nbt.1990

위 논문 "Comprehensive analysis of kinase inhibitor selectivity"에서 활용된 데이터셋은 "Davis dataset"을 활용할 것이다. 위 논문은 2011년, Nature Biotechnology에 발표된 것으로, 다양한 protein kinase inhibitor의 선택성을 광범위하게 분석한 내용을 담고 있다. 논문에서 사용된 데이터는 여러 kinase inhibitor와 다양한 kinase 간의 결합 친화도(affinity)를 측정한 것인데, 이 데이터셋이 매우 유용하고 널리 사용되면서 "Davis dataset"이라는 이름으로 알려지게 되었다. 이 데이터 셋은 약물(또는 억제제)과 타겟 단백질 간의 결합 친화도 데이터를 포함하고 있으며, 실습을 위해 Affinity data, target data, SMILES data로 나누어 사용할 것이다.
Affinity Data(결합 친화도 데이터)
- 내용: 이 데이터는 약물(또는 억제제)과 타겟 단백질 간의 결합 친화도를 나타내는 값으로 구성된다.
- 형식: 일반적으로 Kd (dissociation constant, 해리 상수) 값으로 표현된다. Kd 값이 작을수록 억제제와 타겟 단백질 간의 결합이 강하다는 것을 의미한다.
- 데이터 사용: 모델 학습에서 이 값은 종속 변수로 사용되며, 모델은 주어진 억제제와 타겟 단백질 쌍의 결합 친화도를 예측하도록 훈련된다.


Target Data (타겟 데이터)
- 내용: 이 데이터는 실험에서 사용된 타겟 단백질에 대한 정보를 포함하고 있다.
- 형식: 주로 단백질 서열 정보(아미노산 서열)로 표현되거나, 단백질의 구조적 특징 또는 기능적 특징으로 나타날 수 있다.
- 데이터 사용: 모델은 이 데이터를 사용하여 특정 단백질의 구조적 또는 서열적 특징을 학습하고, 이에 기반하여 예측을 수행한다.
SMILES Data (SMILES 데이터)
- 내용: 이 데이터는 약물(또는 억제제)의 화학적 구조를 기술하는 데 사용된다.
- 형식: SMILES (Simplified Molecular Input Line Entry System)는 분자의 구조를 텍스트 형식으로 표현한 문자열이다. 예를 들어, 벤젠의 경우 SMILES 표기법은 "C1=CC=CC=C1"과 같다.
- 데이터 사용: 모델은 SMILES 데이터를 사용하여 화합물의 화학적 특성을 학습하고, 이러한 화학적 구조가 단백질과의 결합 친화도에 어떻게 영향을 미치는지를 예측한다.
데이터 전처리
데이터 프레임 만들기
데이터 전처리의 목적은 결합 친화도를 이진화하는 것이다. 회귀 문제(결합 친화도를 예측하는 문제)보다 이진 분류 문제는 더 단순하고, 특정 임계값(threshold)을 기준으로 분류하여 결합 여부를 판단하는 실용적인 접근 방식이다. 따라서 SMILES(화합물), Target Sequence(단백질 서열), Affinity(결합 친화도)를 모델이 학습할 수 있는 형식으로 변환하고, 이진 분류 문제로 해석할 것이다.

SMILES - Target - Affinity paired list 만들기
약물(또는 억제제)과 타겟 단백질 쌍을 만들고, 각각의 결합 친화도 값을 저장한다. 데이터셋에는 여러 약물(drug)과 타겟 단백질(target) 쌍이 존재하며, 각각의 결합 친화도(affinity) 정보가 들어있다. 이를 paired list( affinity.values[i, j] )로 만들어서 모델이 학습할 수 있는 형태로 변환할 것이다.
결합 친화도 이진화하기
결합 친화도(affinity)는 보통 Kd 값으로 측정되며, 이 값이 작을수록 약물과 단백질 간 결합이 강하다. 이 코드에서는 Kd <300nM 일 때 결합이 강하다고 판단하여 1로 표시하고, 그 외에는 0으로 표시한다. 이렇게 함으로써 결합 강도에 따라 이진 분류 문제를 설정하여, 모델이 주어진 약물과 단백질이 강하게 결합할지를 예측하도록 할 수 있다.

하나의 데이터 프레임 만들기
약물(SMILES), 타겟 단백질 서열(Target Sequence), 결합 여부(Label)를 하나의 데이터프레임으로 묶어 모델에 입력하기 위한 데이터를 준비한다. 약물의 SMILES sequence, Target portein sequence, 그리고 이진화된 결합 친화도(y)를 하나의 데이터 프레임으로 정리하고 모델 학습에 필요한 입력 데이터와 라벨을 일관성 있게 관리한다.
데이터 분석
전체 pair 수

준비한 전체 데이터 프레임의 row 수는 30056이다.
Label 비율 확인
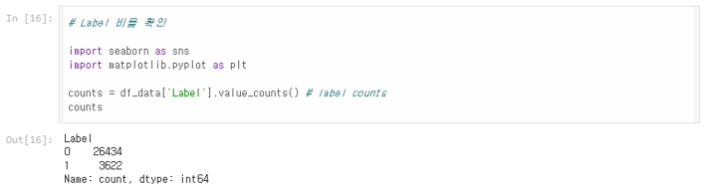

데이터 전처리에서 이진화한 결합 친화도(label)는 0(결합이 약함)이 26434건, 1(결합이 강함)이 3622건이다.
데이터 정제
모델 학습에 활용할 수 있도록 데이터를 정제하는 과정이 필요하다. 흐름을 하나하나 짚어가며 따라가 보자.
아미노산 및 SMILES 문자 정의
# 아미노산 charater 정의
amino_char = ['?', 'A', 'C', 'B', 'E', 'D', 'G', 'F', 'I', 'H', 'K', 'M', 'L', 'O',
'N', 'Q', 'P', 'S', 'R', 'U', 'T', 'W', 'V', 'Y', 'X', 'Z']
# SMILES character 정의
smiles_char = ['?', '#', '%', ')', '(', '+', '-', '.', '1', '0', '3', '2', '5', '4',
'7', '6', '9', '8', '=', 'A', 'C', 'B', 'E', 'D', 'G', 'F', 'I',
'H', 'K', 'M', 'L', 'O', 'N', 'P', 'S', 'R', 'U', 'T', 'W', 'V',
'Y', '[', 'Z', ']', '_', 'a', 'c', 'b', 'e', 'd', 'g', 'f', 'i',
'h', 'm', 'l', 'o', 'n', 's', 'r', 'u', 't', 'y']아미노산 서열(단백질)과 SMILES 문자열에서 허용되는 문자 집합을 정의한다. 이 집합을 통해 데이터에 포함된 문자가 유효한지 검증하고, 유효하지 않거나 불분명한 문자는 ‘?’로 변환한다.
One-Hot 인코더 정의
# protein, drug 원핫(one-hot) 인코더
enc_protein = OneHotEncoder().fit(np.array(amino_char).reshape(-1, 1))
enc_drug = OneHotEncoder().fit(np.array(smiles_char).reshape(-1, 1))아미노산 서열과 SMILES 문자열을 원핫 인코딩할 수 있도록 인코더를 정의한다. OneHotEncoder는 각 문자를 이진 벡터로 변환하여 컴퓨터가 이해할 수 있는 형태로 만든다. 예를 들어, 아미노산 ‘A’는 [1, 0, 0, … ]으로 변환되며, 각 문자가 고유한 이진 벡터를 갖게 된다.
최대 길이 설정
# Protein 최대 길이
MAX_SEQ_PROTEIN = 1000
# Drug 최대 길이
MAX_SEQ_DRUG = 100
SMILES 및 단백질 서열의 길이를 일정하게 맞추기 위한 최대 길이를 설정한다. 서열의 길이가 일정하지 않으면 모델에 입력할 수 없으므로, 일정한 길이로 맞춰야 한다. 만약 서열이 너무 짧으면 뒤에 '?'로 패딩(padding)하고, 너무 길면 최대 길이만큼 자른다.
SMILES 전처리 함수 trans_drug
def trans_drug(x):
"""SMILES 데이터 전처리하기
같은 크기(MAX_SEQ_DRUG)의 리스트로 만들기
사전 정의된 character에 해당 되지 않는 값은 ?로 변환
Args:
x: 하나의 SMILES 데이터 (string)
Return:
전처리된 SMILES 데이터 리스트
"""
temp = list(x) # str -> list
temp = [i if i in smiles_char else '?' for i in temp] # 사전 정의된 character에 없으면 ?로 변환
if len(temp) < MAX_SEQ_DRUG:
temp = temp + ['?'] * (MAX_SEQ_DRUG-len(temp)) # 부족한 부분 ?로 패딩
else:
temp = temp[:MAX_SEQ_DRUG] # 길면 자르기
return temp위 함수는 SMILES 문자열을 일정한 길이와 형식으로 변환하는 함수이다. temp = list(x)는 문자열을 문자 리스트로 변환하고 리스트 내에서 허용되지 않는 문자는 '?'로 변환하며 서열이 MAX_SEQ_DRUG보다 짧으면 '?'로 패딩하고, 길면 앞에서부터 자른다.
Protein 전처리 함수 trans_protein
def trans_protein(x):
"""Protein sequence 데이터 전처리하기
같은 크기(MAX_SEQ_PROTEIN)의 리스트로 만들기
사전 정의된 character에 해당 되지 않는 값은 ?로 변환
Args:
x: 하나의 sequence 데이터 (string)
Return:
전처리된 SMILES 데이터 리스트
"""
temp = list(x.upper()) # 대문자로 변환
temp = [i if i in amino_char else '?' for i in temp] # 허용되지 않은 문자는 ?로
if len(temp) < MAX_SEQ_PROTEIN:
temp = temp + ['?'] * (MAX_SEQ_PROTEIN-len(temp)) # 부족하면 패딩
else:
temp = temp [:MAX_SEQ_PROTEIN] # 길면 자르기
return temp
위 함수는 단백질 서열을 일정한 길이와 형식으로 변환하는 함수이다. 서열을 대문자로 변환한 후 허용되지 않는 아미노산 문자를 '?'로 변환하며 서열이 MAX_SEQ_PROTEIN보다 짧으면 '?'로 패딩하고, 길면 자른다.
전처리 함수 적용
# Drug 데이터 중 중복되지 않는 것만 processing
unique_drug = pd.Series(df_data["SMILES"].unique()).apply(trans_drug)
# raw SMILES - processed SMILES 딕셔너리
unique_dict = dict(zip(df_data["SMILES"].unique(), unique_drug))
# 전체 데이터 processing
df_data["drug_encoding"] = [unique_dict[i] for i in df_data["SMILES"]]
# Protein 데이터 중 중복되지 않는 것만 processing
AA = pd.Series(df_data["Target Sequence"].unique()).apply(trans_protein)
# raw protein - processed protein 딕셔너리
AA_dict = dict(zip(df_data["Target Sequence"].unique(), AA))
# 전체 데이터 processing
df_data["target_encoding"] = [AA_dict[i] for i in df_data["Target Sequence"]]
중복되지 않는 SMILES 데이터와 protein 데이터에 대해서 전처리 함수를 적용하고 원본과 처리된 데이터를 매핑한 딕셔너리를 생성한 후 전체 데이터에 적용한다. 이로써 SMILES와 단백질 서열 데이터를 일정한 길이로 맞추고 유효하지 않은 문자를 처리하여 학습 가능한 형태로 정제하는 과정을 수행했다. 각각의 서열은 원핫 인코딩 준비를 위해 문자열에서 문자 리스트로 변환되며, 모든 서열이 동일한 길이가 되도록 처리되었다.

데이터셋 나누기
이제 모델의 훈련을 위해 데이터셋을 나누는 과정을 거친다.
def create_fold(df, fold_seed, frac):
"""Train / Val / Test 나누기
Args:
df: 전체 데이터 (Pandas DataFrame)
fold_seed: random_state for random sampling
frac: train - val - test fraction (list or tuple) ex) [0.7,0.1,0.2]
Returns:
Train / Val / Test dataframe (tuple)
"""
train_frac, val_frac, test_frac = frac
# Test 데이터 random sampling
test = df.sample(frac = test_frac, replace = False, random_state = fold_seed)
train_val = df[~df.index.isin(test.index)] # Train & Validation 데이터
# Validation 데이터 random sampling
val = train_val.sample(frac = val_frac/(1-test_frac), replace = False, random_state = 1)
train = train_val[~train_val.index.isin(val.index)] # Train 데이터
return train, val, test
위 함수는 데이터를 Train(학습용), Validation(검증용), Test(테스트용) 세 개의 데이터 셋으로 나누기 위한 함수이다. 데이터 샘플링 시 사용하는 fold_seed(random_state)와 세 개의 데이터 셋의 비율을 나타내는 frac을 파라미터로 받는다.
그 후, df.sample()을 사용하여 전체 데이터의 정해진 frac만큼 무작위로 샘플링한다.
create_fold 적용
train, val, test = create_fold(df_data, 22, [0.7,0.1,0.2])
# 잘 나누어졌는지 확인
print(f'Train 데이터 크기: {len(train)}')
print(f'Validation 데이터 크기: {len(val)}')
print(f'Test 데이터 크기: {len(test)}')
# Train 데이터 크기: 21039
# Validation 데이터 크기: 3006
# Test 데이터 크기: 6011
위와 같은 조건으로 create_fold를 수행했고 그 결과 Train 데이터 21039건, Validation 데이터 3006건, Test 데이터 6011건으로 분할되었다.

댓글